There has been much hype over the new paper published in the latest Nature issue which claims to have discovered an opioid analgesic that doesn’t have most of the side effects of morphine. If the claim holds, the authors may have found the Holy Grail of pain research chased by too many for too long (besides being worth billions of dollars to its discoverers).
The drug, called PZM21, was discovered using structure-based drug design. This means that instead of taking a drug that works, say morphine, and then tweaking its molecular structure in various ways and see if the resultant drugs work, you take the target of the drug, say mu-opioid receptors, and design a drug that fits in that slot. The search and design are done initially with sophisticated software and there are many millions of virtual candidates. So it takes a lot of work and ingenuity to select but a few drugs that will be synthesized and tested in live animals.
Manglik et al. (2016) did just that and they came up with PZM21 which, compared to morphine, is:
1) selective for the mu-opioid receptors (i.e. it doesn’t bind to anything else)
2) produces no respiratory depression (maybe a touch on the opposite side)
3) doesn’t affect locomotion
4) produces less constipation
5) produces long-lasting affective analgesia
6) and has less addictive liability
The Holy Grail, right? Weeell, I have some serious issues with number 5 and, to some extent, number 6 on this list.
Normally, I wouldn’t dissect a paper so thoroughly because, if there is one thing I learned by the end of GradSchool and PostDoc, is that there is no perfect paper out there. Consequently, anyone with scientific training can find issues with absolutely anything published. I once challenged someone to bring me any loved and cherished paper and I would tear it apart; it’s much easier to criticize than to come up with solutions. Probably that’s why everybody hates Reviewer No. 2…
But, for extraordinary claims, you need extraordinary evidence. And the evidence simply does not support the 5 and maybe 6 above.
Let’s start with pain. The authors used 3 tests: hotplate (drop a mouse on a hot plate for 10 sec and see what it does), tail-flick (give an electric shock to the tail and see how fast the mouse flicks its tail) and formalin (inject an inflammatory painful substance in the mouse paw and see what the animal does). They used 3 doses of PZM21 in the hotplate test (10, 20, and 40 mg/Kg), 2 doses in the tail-flick test (10 and 20), and 1 dose in the formalin test (20). Why? If you start with a dose-response in a test and want to convince me it works in the other tests, then do a dose-response for those too, so I have something to compare. These tests have been extensively used in pain research and the standard drug used is morphine. Therefore, the literature is clear on how different doses of morphine work in these tests. I need your dose-responses for your new drug to be able to see how it measures up, since you claim it is “more efficacious than morphine”. If you don’t want to convince me there is a dose-response effect, that’s fine too, I’ll frown a little, but it’s your choice. However, then choose a dose and stick with it! Otherwise I cannot compare the behaviors across tests, rendering one or the other test meaningless. If you’re wondering, they used only one dose of morphine in all the tests, except the hotplate, where they used two.
Another thing also related to doses. The authors found something really odd: PZM21 works (meaning produces analgesia) in the hotplate, but not the tail-flick tests. This is truly amazing because no opiate I know of can make such a clear-cut distinction between those two tests. Buuuuut, and here is a big ‘BUT” they did not test their highest dose (40mg/kg) in the tail-flick test! Why? I’ll tell you how, because I am oh sooo familiar with this argument. It goes like this:
Reviewer: Why didn’t you use the same doses in all your 3 pain tests?
Author: The middle and highest doses have similar effects in the hotplate test, ok? So it doesn’t matter which one of these doses I’ll use in the tail-flick test.
Reviewer: Yeah, right, but, you have no proof that the effects of the two doses are indistinguishable because you don’t report any stats on them! Besides, even so, that argument applies only when a) you have ceiling effects (not the case here, your morphine hit it, at any rate) and b) the drug has the expected effects on both tests and thus you have some logical rationale behind it. Which is not the case here, again: your point is that the drug DOESN’T produce analgesia in the tail-flick test and yet you don’t wanna try its HIGHEST dose… REJECT AND RESUBMIT! Awesome drug discovery, by the way!
So how come the paper passed the reviewers?! Perhaps the fact that two of the reviewers are long term publishing co-authors from the same University had something to do with it, you know, same views predisposes them to the same biases and so on… But can you do that? I mean, have reviewers for Nature from the same department for the same paper?
Alrighty then… let’s move on to the stats. Or rather not. Because there aren’t any for the hotplate or tail-flick! Now, I know all about the “freedom from the tyranny of p” movement (that is: report only the means, standard errors of mean, and confidence intervals and let the reader judge the data) and about the fact that the average scientist today needs to know 100-fold more stats that his predecessors 20 years ago (although some biologists and chemists seem to be excused from this, things either turn color or not, either are there or not etc.) or about the fact that you cannot get away with only one experiment published these days, but you need a lot of them so you have to do a lot of corrections to your stats so you don’t fall into the Type 1 error. I know all about that, but just like the case with the doses, choose one way or another and stick to it. Because there are ANOVAs ran for the formalin test, the respiration, constipation, locomotion, and conditioned place preference tests, but none for the hotplate or tailflick! I am also aware that to be published in Science or Nature you have to strip your work and wordings to the bare minimum because the insane wordcount limits, but you have free rein in the Supplementals. And I combed through those and there are no stats there either. Nor are there any power analyses… So, what’s going on here? Remember, the authors didn’t test the highest dose on the tail-flick test because – presumably – the highest and intermediary doses have indistinguishable effects, but where is the stats to prove it?
And now the thing that really really bothered me: the claim that PZM21 takes away the affective dimension of pain but not the sensory. Pain is a complex experience that, depending on your favourite pain researcher, has at least 2 dimensions: the sensory (also called ‘reflexive’ because it is the immediate response to the noxious stimulation that makes you retract by reflex the limb from whatever produces the tissue damage) and the affective (also called ‘motivational’ because it makes the pain unpleasant and motivates you to get away from whatever caused it and seek alleviation and recovery). The first aspect of pain, the sensory, is relatively easy to measure, since you look at the limb withdrawal (or tail, in the case of animals with prolonged spinal column). By contrast, the affective aspect is very hard to measure. In humans, you can ask them how unpleasant it is (and even those reports are unreliable), but how do you do it with animals? Well, you go back to humans and see what they do. Humans scream “Ouch!” or swear when they get hurt (so you can measure vocalizations in animals) or humans avoid places in which they got hurt because they remember the unpleasant pain (so you do a test called Conditioned Place Avoidance for animals, although if you got a drug that shows positive results in this test, like morphine, you don’t know if you blocked the memory of unpleasantness or the feeling of unpleasantness itself, but that’s a different can of worms). The authors did not use any of these tests, yet they claim that PZM21 takes away the unpleasantness of pain, i.e. is an affective analgesic!
What they did was this: they looked at the behaviors the animal did on the hotplate and divided them in two categories: reflexive (the lifting of the paw) and affective (the licking of the paw and the jumping). Now, there are several issues with this dichotomy, I’m not even going to go there; I’ll just say that there are prominent pain researchers that will scream from the top of their lungs that the so-called affective behaviors from the hotplate test cannot be indexes of pain affect, because the pain affect requires forebrain structures and yet these behaviors persist in the decerebrated rodent, including the jumping. Anyway, leaving the theoretical debate about what those behaviors they measured really mean aside, there still is the problem of the jumpers: namely, the authors excluded from the analysis the mice who tried to jump out of the hotplate test in the evaluation of the potency of PZM21, but then they left them in when comparing the two types of analgesia because it’s a sign of escaping, an emotionally-valenced behavior! Isn’t this the same test?! Seriously? Why are you using two different groups of mice and leaving the impression that is only one? And oh, yeah, they used only the middle dose for the affective evaluation, when they used all three doses for potency…. And I’m not even gonna ask why they used the highest dose in the formalin test… but only for the normal mice, the knockouts in the same test got the middle dose! So we’re back comparing pears with apples again!
Next (and last, I promise, this rant is way too long already), the non-addictive claim. The authors used the Conditioned Place Paradigm, an old and reliable method to test drug likeability. The idea is that you have a box with 2 chambers, X and Y. Give the animal saline in chamber X and let it stay there for some time. Next day, you give the animal the drug and confine it in chamber Y. Do this a few times and on the test day you let the animal explore both chambers. If it stays more in chamber Y then it liked the drug, much like humans behave by seeking a place in which they felt good and avoiding places in which they felt bad. All well and good, only that is standard practice in this test to counter-balance the days and the chambers! I don’t know about the chambers, because they don’t say, but the days were not counterbalanced. I know, it’s a petty little thing for me to bring that up, but remember the saying about extraordinary claims… so I expect flawless methods. I would have also liked to see a way more convincing test for addictive liability like self-administration, but that will be done later, if the drug holds, I hope. Thankfully, unlike the affective analgesia claims, the authors have been more restrained in their verbiage about addiction, much to their credit (and I have a nasty suspicion as to why).
I do sincerely think the drug shows decent promise as a painkiller. Kudos for discovering it! But, seriously, fellows, the behavioral portion of the paper could use some improvements.
Ok, rant over.
EDIT (Aug 25, 2016): I forgot to mention something, and that is the competing financial interests declared for this paper: some of its authors already filed a provisional patent for PZM21 or are already founders or consultants for Epiodyne (a company that that wants to develop novel analgesics). Normally, that wouldn’t worry me unduly, people are allowed to make a buck from their discoveries (although is billions in this case and we can get into that capitalism-old debate whether is moral to make billions on the suffering of other people, but that’s a different story). Anyway, combine the financial interests with the poor behavioral tests and you get a very shoddy thing indeed.
Reference: Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Da Duan, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, & Shoichet BK (Epub 17 Aug 2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature, 1-6. PMID: 27533032, DOI: 10.1038/nature19112. ARTICLE
By Neuronicus, 21 August 2016

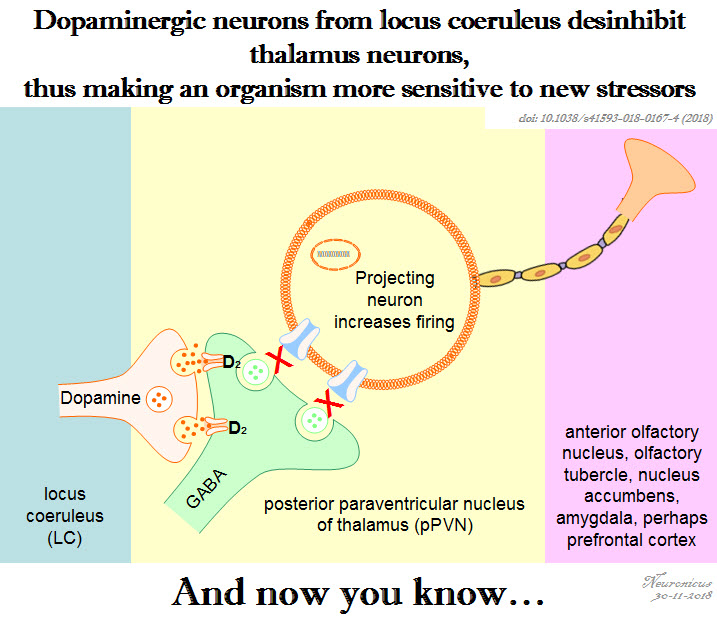







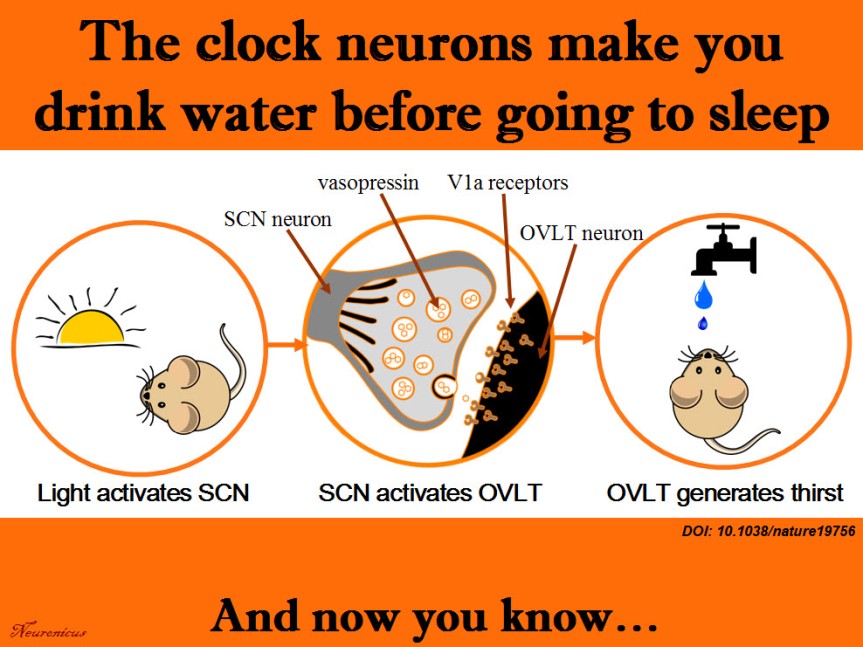








 Every neuroscience or biology textbook will tell you that neurons communicate with one another in two ways: via chemical synapses (one neuron releases some substances that change the membrane potential of the receiving neuron) or via electrical synapses (neurons physically share some membrane proteins that allows the electrical impulse to go from one neuron to another). (Nota bene: for the taxonomic nitpickers, I decided that the other non-conventional forms of communication, like diffusion, fall in one or the other of the two categories above).
Every neuroscience or biology textbook will tell you that neurons communicate with one another in two ways: via chemical synapses (one neuron releases some substances that change the membrane potential of the receiving neuron) or via electrical synapses (neurons physically share some membrane proteins that allows the electrical impulse to go from one neuron to another). (Nota bene: for the taxonomic nitpickers, I decided that the other non-conventional forms of communication, like diffusion, fall in one or the other of the two categories above).
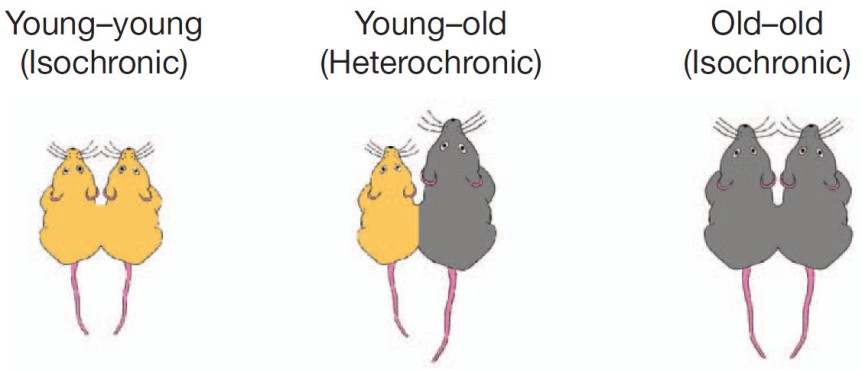



 When you’re sick you also feel awful: no appetite, weak, sleepy, feverish, achy, and so on. This is called, appropriately so, the sickness syndrome.
When you’re sick you also feel awful: no appetite, weak, sleepy, feverish, achy, and so on. This is called, appropriately so, the sickness syndrome.















